
|
||||||||||
|
||||||||||



SUPPORT/Methods of nucleic acid research:

|
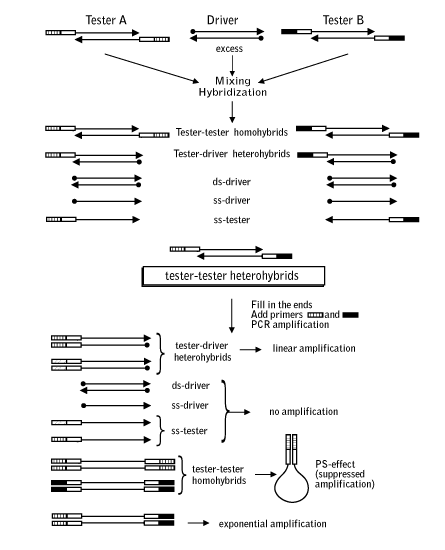
Suppression Subtractive Hybridization (SSH)Subtractive hybridization is an approach that allows comparison of two DNA populations and isolation of a fraction enriched in differentially distributed molecules. Subtractive hybridization is usually employed to identify genes with a differential expression pattern, in particular genes involved in the regulation of basic biological processes. Suppression Subtractive Hybridization (SSH) is the best known and most utilized subtraction method (Lukyanov et al., 1994; Gurskaya et al., 1996; Diachenko et al., 1996). It is based on the PCR suppression by inverted terminal repeats (PS-effect). Upon initiation of PCR after the denaturation phase, the single-stranded (ss) DNA fragments flanked by inverted terminal repeats (ITR) may form either self-annealing "pan-like" structures (preventing the primer binding to its complementary binding sites and suppressing the PCR), or DNA/primer hybrid structures. In the latter case, if the primer corresponding to the outer part of ITR is used, the DNA synthesis by Taq-polymerase restores the original structure, ensuring the persistence of suppression during further PCR cycles. In complex mixtures, the use of PCR suppression allows selective amplification of molecules that are flanked by different adapters at opposing termini (asymmetrically flanked molecules).
This principle is used in SSH, where the molecules of interest are driven to the asymmetrically flanked state, leaving the ballast fraction symmetrically flanked (i.e. without sites for primer annealing). The substrate for SSH is thermally denatured double-stranded (ds) cDNA (the tester) containing specifically expressed sequences to be extracted (the target), and denatured ds-cDNA lacking the target sequence(s) (the driver) used for comparison. Tester and driver cDNA samples are digested with a blunt cutting restriction enzyme, then the resulting tester cDNA fragments are subdivided into two samples (A and B) and ligated to the corresponding different "pseudo-double-stranded" adapters A and B at their 5' ends. During the first stage of subtraction, excess driver is added to each sample of tester and the samples are denatured and allowed to re-anneal. In this step, tester molecules corresponding to the driver (termed from this point "redundant molecules") form driver-tester heterohybrids removing them from the ss-fraction. Due to the second-order kinetics of the hybridization reaction, it is highly effective for the abundant redundant molecules, leading to the approximate equalization of concentrations of various types of redundant molecules. Meanwhile, target molecules are not affected by "driver pressing" and the remaining ss-tester becomes target enriched. Since target molecules form homohybrids with each other (reassociate), and since reassociation progresses much more rapidly for abundant targets than for rare sequences, the concentration of target molecules in the ss-tester is equalized. During the second stage of subtraction, samples A and B (having different ligated adaptors) are mixed and allowed to reassociate. This leads to the creation of novel molecules from the subtracted ss-DNA that are by definition asymmetrically flanked by adapter A at one end and adaptor B at the other. These molecules may now be isolated by PS-effect PCR using primers that recognize the outer parts of adapters A and B. Amplification of symmetrically flanked testers from the first stage will be suppressed, and hybrids formed with the driver will also not amplify due to the lack of one or both primer annealing sites, leading to preferential amplification of the asymmetrically flanked sequences of interest.
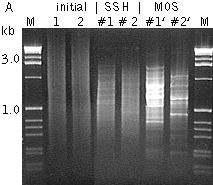
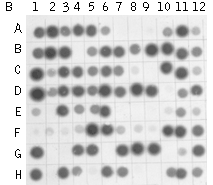
Mirror Orientation Selection SSH method has been applied in many hundreds of models. Due to high efficiency of this technology, unwanted background clones representing non-differentially expressed transcripts can be generated by SSH. First, unincorporated adapters can randomly anneal to non-target cDNA molecules during subtractive hybridization. After DNA elongation, such molecules can serve as a template for amplification and will be picked up in the final product, as will product generated by non-specific annealing of PCR primers. The second type of the background is generated from very abundant redundant sequences. Redundant cDNA molecules can evade elimination by hybridization with driver and will amplify in subsequent PCR steps. For any given redundant cDNA, this is extremely unlikely. In most cases just a single molecule of each redundant cDNA will exist among several thousand other cDNA molecules after the subtractive hybridization and will not amplify as a false positive. However, in the case of a very abundant sequence, sufficient excess can be present to allow amplification in subsequent steps. Clones representing type I background can be identified by differential screening because they do not produce a differential signal. On the contrary, type II background clones show differential signals during screening with probes prepared from two reciprocal (forward and reverse) subtracted samples. Only Northern blot and RT-PCR analysis can demonstrate the equal abundance of such sequences in the initial mRNA samples. Therefore, elimination of this background is a difficult and time-consuming step in subtracted library analysis. As an alternative to screening with subtracted probes, it is possible to use the unsubtracted tester and driver cDNA as probes for differential screening, but in this case many clones representing rare transcripts give no signals. The Mirror Orientation Selection (MOS) method has been recently developed to eliminate type II background from SSH-generated libraries (Rebrikov et al., 2000).
Subtraction of bacterial genomes SSH method has been adapted for comparison of bacterial genomes (Akopyants et al., 1998). Genes that are present in certain isolates of a given bacterial species and absent or substantially different in others can be of great interest biologically. Some may determine strain-specific traits such as drug resistance, bacterial surface structure, or restriction-modification. Of special importance in infectious disease are the "pathogenicity islands" (PAIs), multigene segments of virulent strains that tend to be absent from avirulent members of the same species and that help determine the nature and severity of disease. To select strain-specific genes in bacterial genomes using SSH, pools of digested genomic DNA from the strain of interest (tester) are deprived of sequences also present in the reference strain (driver) by judicious use of hybridization and PCR. The remaining tester-enriched DNA fragments are then cloned for further analysis. References
|
||||||||||||


|
Copyright 2002-2023 Evrogen. All rights reserved. Evrogen JSC, 16/10 Miklukho-Maklaya str., Moscow, Russia, Tel +7(495)988-4084, Fax +7(495)988-4085, e-mail:evrogen@evrogen.com |